
The personalized editing therapy developed at record speed for the American newborn required preliminary studies, favorable circumstances, and a heroic collective effort.
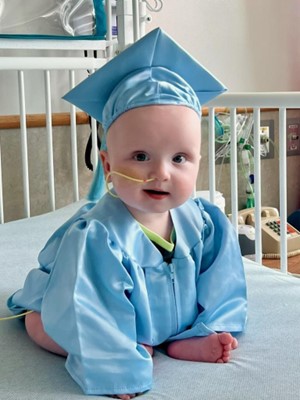
After 307 days, little KJ Muldoon was discharged from the Children’s Hospital of Philadelphia (CHOP), wearing a tiny graduation outfit complete with a blue gown and cap. Born ten months ago with a severe metabolic disorder, the baby received a genome editing therapy developed exclusively for him, and his remarkable progress has been hailed by many as the dawn of a new era in precision medicine. A month after the publication of his case in the New England Journal of Medicine, we take a closer look at how researchers managed to develop the treatment in just six months—and whether this breakthrough could be replicated for other rare disease patients in need of life-saving therapies.
The key element was the extraordinary teamwork that made this feat possible. Under normal conditions, creating and delivering a tailored therapy takes years, time that many fast-progressing diseases don’t allow. To compress the research, development, production, and authorization timeline, a dream team had to perform a kind of Olympic relay. Much of the credit goes to CHOP and its affiliated university (the University of Pennsylvania) especially metabolic disease specialist Rebecca Ahrens-Nicklas and genome editing pioneer Kiran Musunuru. They had previously worked on a mouse model for another metabolic disorder (phenylketonuria), which proved to be invaluable experience when the ideal human case arrived: a patient with carbamoyl phosphate synthetase 1 (CPS1) deficiency.
They were joined by some of the best in the field: the Innovative Genomics Institute (IGI), founded by CRISPR co-inventor Jennifer Doudna; the Jackson Laboratory, a nonprofit with international reach; and three biotech companies: Aldevron and Integrated DNA Technologies (both part of the Danaher Corporation), and Acuitas Therapeutics. United by the goal of breaking records, the team worked to go from CRISPR drug design to patient delivery as fast as possible. The details of this collaboration, also enabled by the foresight of the National Institutes of Health, were shared on the IGI website.
KJ was born on August 1, 2024, and doctors in Philadelphia quickly identified a condition so rare it affects only one newborn in 1.3 million. At that moment, the network of ideas and relationships needed for the challenge was already in place and ready to spring into action. Within days, Musunuru — previously known mostly for his cardiovascular research —contacted another editing pioneer focused on rare diseases: Fyodor Urnov, who works at IGI to accelerate the translation of CRISPR therapies from the lab to the bedside.
When Urnov learned of the baby with CPS1 deficiency — a condition with a known genetic cause but no cure — he thought: “This is what we’ve been preparing for our entire lives.” He reached out to Danaher partners working with IGI on the Beacon for CRISPR Cures project, which is currently focused on two inherited immune disorders (ART-SCID and HLH). His message: “We have to do this.” The Jackson Laboratory promptly joined, tasked with providing custom mouse models to test the therapy’s safety and efficacy.
KJ and his parents received full genome sequencing in record time, thanks to another IGI initiative called Ingenuiti. Despite his illness, KJ was fortunate in two ways: his genetic defect consisted of a single-letter error on the DNA, perfect for base editing, and it affected the liver, an organ well-suited to molecular delivery thanks to lipid nanoparticles. Knowing KJ’s full genome also helped researchers assess and minimize the risks of off-target effects.
As soon as Musunuru and his team at the University of Pennsylvania designed the guide molecule to correct the typo in the DNA, IGI began safety testing to prepare the data needed for regulatory approval. Biotech companies played a crucial role in producing therapeutic components under clinical standards. Everyone worked under intense pressure, even through the Christmas holidays.
Ultimately, the Food and Drug Administration took just one week to authorize this highly personalized therapy, which, given the rarity of the condition, may never be used for another patient. KJ received a very low initial dose on February 25. After confirming there were no adverse effects, the team administered a stronger dose in March and another in April. For ethical reasons, they avoided a liver biopsy (which would have confirmed DNA repair). Still, the indirect evidence is compelling: KJ has resumed a normal diet, is growing well, has greatly reduced his medication to prevent ammonia buildup in his blood, and no longer needs a liver transplant. He’s not cured, but if all goes well, his condition will be much milder and manageable.
Those behind this scientific achievement are energized by the results, as reflected in comments gathered by IGI, Inside Precision Medicine, and STAT:
“This is a landmark moment for CRISPR in medicine, and we can never look back. It can be done even faster. Now we have to show that we can scale this from CRISPR for one to CRISPR for all” (F. Urnov).
“I’m overwhelmed to see the pictures of KJ at home, with his family, for the first time. Somehow that hits harder, makes it more real for me, than anything else that’s happened over the past 10 months. And it just increases my determination to find ways to do the same for as many kids as possible” (K. Musunuru).
“It was a remarkable team effort. The ability to develop an on-demand CRISPR therapy in such a short time opens up a new era for treating previously untreatable genetic diseases” (J. Doudna).
No one knows exactly how much this largely volunteer-based public-private effort cost, and unfortunately, it’s unlikely that such a heroic undertaking can be repeated over and over for every rare or ultra-rare disease. But the proof of principle is there. The next step is figuring out how to make custom editing therapies a little less exceptional, through technological advances and systemic reforms.
Jason Mast covered the annual meeting of the American Society of Gene and Cell Therapy for STAT, where KJ’s data was presented on May 15, and captured the mood among top specialists in the field. “Depending on who you asked, either that was sort of a one-off, that is a nice ray of sunshine that’s not going to be repeated, or it’s a part of the future of gene editing and what this tech can do,” he said on the publication’s podcast. He also recalled that the sector has lately been clouded by investor fatigue and the challenges of bringing experimental therapies into the real world. We don’t yet know if or when this shining exception will become the rule, but KJ represents a hope that many researchers and patients truly needed.
(translated and adapted from an article by Anna Meldolesi in Osservatorio Terapie Avanzate)
